








Every cell in the human body is packed with tiny organelles called mitochondria (Fig. 1). Mitochondria are often referred to as “intracellular powerhouses” because they produce most of the energy used by the body. Cells with a high metabolic rate, such as heart muscle cells, may be equipped with thousands of mitochondria, while other cells may only contain a few dozen.
Mitochondrial energy production is an absolute necessity for physical strength, energy, stamina, and life itself. Even the slightest drop in mitochondrial energy output, no matter how subtle, can lead to weakness, fatigue and cognitive difficulties. Unfortunately, during aging, mitochondrial function—like just about everything else—becomes less efficient.
 Mitochondrial Energy Production
Mitochondrial Energy Production
Mitochondrial energy production is accomplished by two closely linked metabolic processes: (1) the citric acid cycle (also known as the Krebs’, or tricarboxylic acid cycle), and (2) oxidative phosphorylation (OXPHOS).
The citric acid cycle (Fig. 2) converts biological fuel (carbohydrates and fats) into adenosine triphosphate (ATP). ATP is the body’s principal source of cellular energy.
 The other process—oxidative phosphorylation (OXPHOS)—combines hydrogen with oxygen to generate even larger amounts of ATP. OXPHOS generates about ten times more ATP than the citric acid cycle. Oxidative phosphorylation is the primary energy process for all aerobic organisms.
The other process—oxidative phosphorylation (OXPHOS)—combines hydrogen with oxygen to generate even larger amounts of ATP. OXPHOS generates about ten times more ATP than the citric acid cycle. Oxidative phosphorylation is the primary energy process for all aerobic organisms.
Mitochondrial oxidative phosphorylation (OXPHOS) provides over 80 percent of the ATP used by our cells for energy. OXPHOS operates very efficiently—close to 70 percent. This compares with about 10 percent for internal combustion engines, and about 40 percent for hydrogen-oxygen fuel cells used in spacecraft.
OXPHOS operates through the electron transport chain (Fig. 3)—a chain of five protein complexes embedded in the inner membrane of the mitochondria. These complexes include:
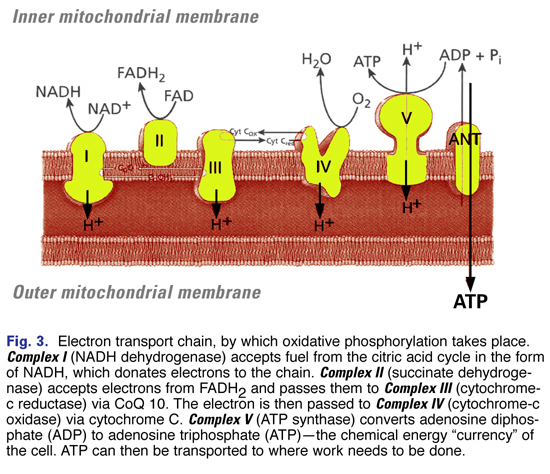
- Complex I (NADH dehydrogenase)
- Complex II (succinate dehydrogenase)
- Complex III (cytochrome-c reductase)
- Complex IV (cytochrome-c oxidase)
- Complex V (ATP synthase)
Oxidative phosphorylation converts adenosine diphosphate (ADP) to adenosine triphosphate (ATP)—the chemical energy “currency” of the cell—that can then be transported to where work needs to be done.
 Age-Related Changes in Mitochondrial Function – A Cause of Aging?
Age-Related Changes in Mitochondrial Function – A Cause of Aging?
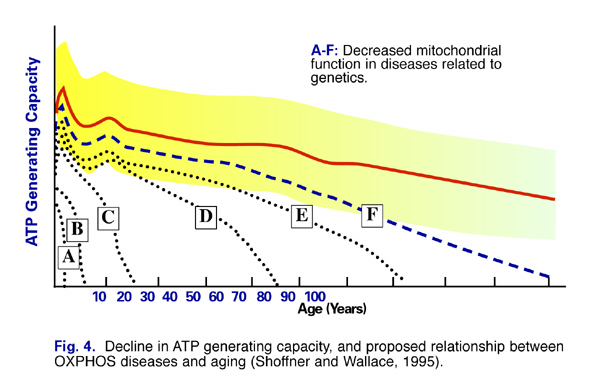
The efficiency of mitochondrial energy production and function declines with age (Fig. 4). Evidence is accumulating that mitochondrial dysfunction underlies many common age-related diseases, including Parkinson’s disease, Alzheimer’s disease, heart disease, fatigue syndromes, and numerous genetic conditions (Fig.5). (1) Dr. Christoph Richter of the Swiss Federal Institute of Technology in Zurich wrote, “A decline in oxidative phosphorylation capacity has been proposed to limit life expectancy (1995).” (2)
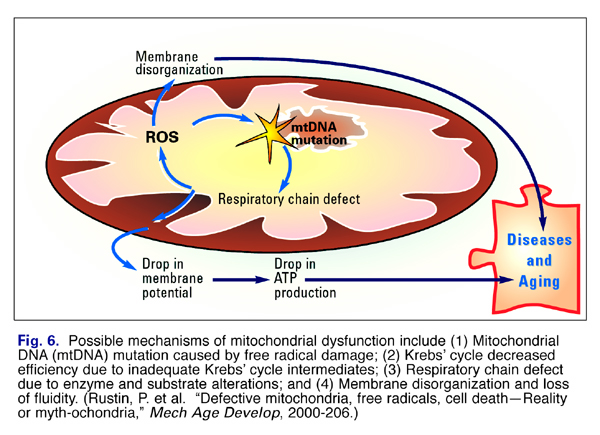
 Changes that occur with aging in mitochondria include (1) damage to mitochondrial DNA by free radicals; (2) decreased efficiency of the Krebs’ cycle; (3) altered response to long term energy demands, (4) Respiratory chain defect due to enzyme alterations; and (5) decreased membrane fluidity (Fig. 6).
Changes that occur with aging in mitochondria include (1) damage to mitochondrial DNA by free radicals; (2) decreased efficiency of the Krebs’ cycle; (3) altered response to long term energy demands, (4) Respiratory chain defect due to enzyme alterations; and (5) decreased membrane fluidity (Fig. 6).
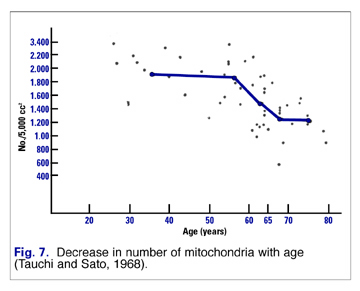 Mitochondria of healthy young adults respond differently to increased requirements for energy than older folks. Mitochondria of youthful people adapt to increased energy requirements by replicating rapidly. This results in more mitochondria producing more ATP for energy. However, as we age, our mitochondria replicate less readily. That means there are fewer mitochondria to produce the energy (Fig. 7).
Mitochondria of healthy young adults respond differently to increased requirements for energy than older folks. Mitochondria of youthful people adapt to increased energy requirements by replicating rapidly. This results in more mitochondria producing more ATP for energy. However, as we age, our mitochondria replicate less readily. That means there are fewer mitochondria to produce the energy (Fig. 7). 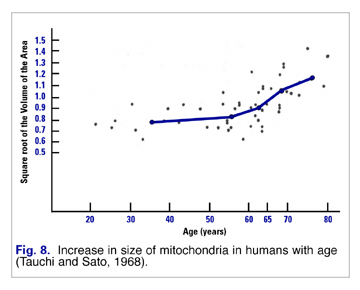
These fewer mitochondria attempt to respond to increased demand by hypertrophying (increasing in size) (Fig. 8). Unfortunately these larger mitochondria are less efficient and produce more damaging free radicals than their more youthful, more numerous, and smaller progenitors.
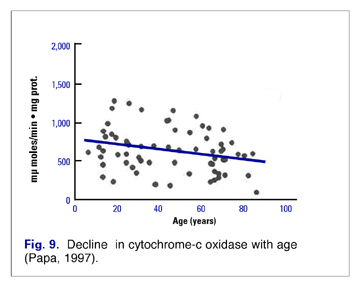 These alterations in mitochondrial efficiency and function are at least partly related to alterations in concentration and efficiency of the elements of the respiratory chain. Most studies indicate a decrease in Complexes I (NADH dehydrogenase), II (succinate dehydrogenase), and IV (Cytochrome-c oxidase) (Fig. 9).(3) In addition, as the amount of mitochondrial enzymes changes with age, the number of cells that totally lack cytochrome oxidase increases. (4)
These alterations in mitochondrial efficiency and function are at least partly related to alterations in concentration and efficiency of the elements of the respiratory chain. Most studies indicate a decrease in Complexes I (NADH dehydrogenase), II (succinate dehydrogenase), and IV (Cytochrome-c oxidase) (Fig. 9).(3) In addition, as the amount of mitochondrial enzymes changes with age, the number of cells that totally lack cytochrome oxidase increases. (4)
Free Radicals as a Cause of Mitochondrial Aging
Dr. Denham Harman first proposed mitochondrial dysfunction as an underlying cause of aging and age-related diseases in 1972 (Dr. Harman related the fascinating history of this discovery in the July, 2002 issue of Vitamin Research News).(5) Following Harman’s lead, other scientists modified or extended his theory. Dr. Jaime Miguel’s Oxygen Stress-Mitochondrial Mutation Theory of Aging; (6-8) Prof. Imre Zs.Nagy’s Membrane Theory of Aging; (9-12) and more recently, Aubrey DeGrey’s Mitochondrial Free Radical Theory of Aging 14and Reductive Hotspot Hypothesis of Aging (15) are all free radical-related theories. It should be noted that none of these theories are mutually exclusive—rather, they complement each other, for the most part.
The theories generally focus on the possible mechanisms by which mitochondria can become dysfunctional and contribute to aging and age-related diseases as shown in the schematic in Fig. 6.
Susceptibility of Mitochondrial DNA to Free Radical Damage
 A unique property of mitochondria is that they have their own DNA (deoxy-ribonucleic acid). DNA is the stuff of which genes and chromosomes are made. Mitochondrial DNA (mtDNA) is quite different from nuclear DNA (nDNA) in several respects. First, mtDNA is not associated with histones. Histones are positively charged “storage” proteins around which nuclear DNA is wound (like thread on a spool) which are vital for the protection of DNA. MtDNA lacks this protective mechanism. Second, most of the complex DNA repair mechanisms that correct damage to nuclear DNA are missing from mitochondria. Therefore, mtDNA lacks both protective and repair mechanisms.
A unique property of mitochondria is that they have their own DNA (deoxy-ribonucleic acid). DNA is the stuff of which genes and chromosomes are made. Mitochondrial DNA (mtDNA) is quite different from nuclear DNA (nDNA) in several respects. First, mtDNA is not associated with histones. Histones are positively charged “storage” proteins around which nuclear DNA is wound (like thread on a spool) which are vital for the protection of DNA. MtDNA lacks this protective mechanism. Second, most of the complex DNA repair mechanisms that correct damage to nuclear DNA are missing from mitochondria. Therefore, mtDNA lacks both protective and repair mechanisms.
 Mitochondrial DNA (mtDNA) is located in the mitochondrial matrix, near the inner mitochondrial membrane (where both energy and free radicals are produced). Since mtDNA lacks protective proteins, it is even more vulnerable to free radical attack.
Mitochondrial DNA (mtDNA) is located in the mitochondrial matrix, near the inner mitochondrial membrane (where both energy and free radicals are produced). Since mtDNA lacks protective proteins, it is even more vulnerable to free radical attack.
 Mitochondrial dysfunction begins with a less-than-perfect electron transport system. Even under ideal conditions, some electrons “leak” from the electron transport chain. These leaking electrons interact with oxygen to produce superoxide radicals. Because mtDNA is close to the site of the production of superoxide, hydroxyl, or perhydroxyl radicals, and because it lacks protective and repair mechanisms, it is highly susceptible to free radical-induced mtDNA deletions (damage). In fact, the relatively unprotected and unrepaired mtDNA suffers more than ten times the damage that nuclear DNA does. (7,8,16)
Mitochondrial dysfunction begins with a less-than-perfect electron transport system. Even under ideal conditions, some electrons “leak” from the electron transport chain. These leaking electrons interact with oxygen to produce superoxide radicals. Because mtDNA is close to the site of the production of superoxide, hydroxyl, or perhydroxyl radicals, and because it lacks protective and repair mechanisms, it is highly susceptible to free radical-induced mtDNA deletions (damage). In fact, the relatively unprotected and unrepaired mtDNA suffers more than ten times the damage that nuclear DNA does. (7,8,16)
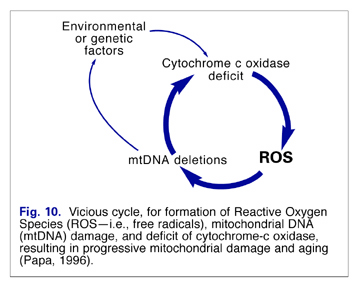
Unfortunately, with mitochondrial dysfunction, leakage of electrons can increase significantly, and a vicious cycle can be created (Fig. 10). These electrons (free radicals) can damage the mitochondrial membrane, resulting in a loss of membrane fluidity and alteration in its permeability. This can lead to further mitochondrial dysfunction, disruption of cellular energy production, and accelerated cellular aging. (6)
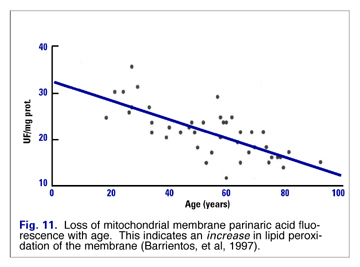
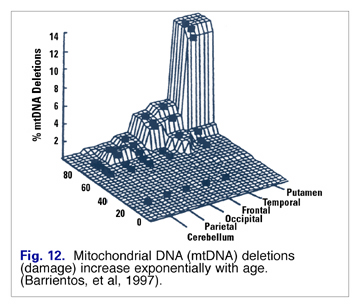
Lipid peroxide damage to mitochondrial membranes increases in a linear fashion (Fig. 11), and mtDNA mutations increase exponentially with age (Fig. 12). Barrientos speculated that changes in membrane lipid composition could be a cause of age-related decreased membrane fluidity. (17)
Approaches to Resuscitate Aging Mitochondria
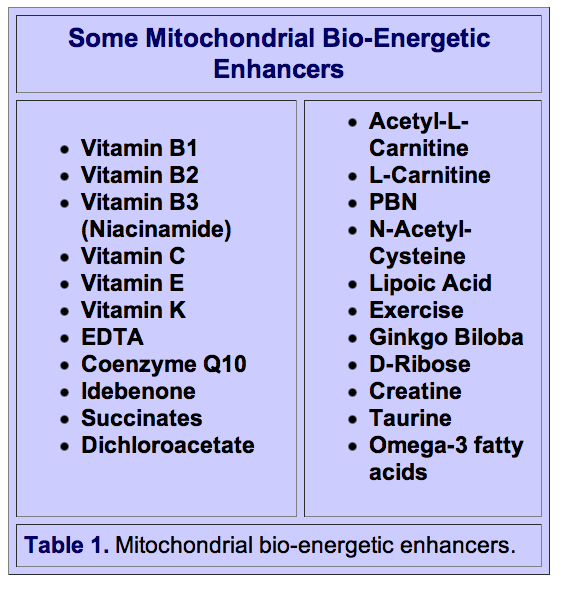
Understanding the proposed mechanisms by which mitochondrial dysfunction can contribute to aging and aging related diseases suggests several potential interventions. These include (1) maintenance of optimal Krebs’ cycle and respiratory chain efficiency, (2) restoration of mitochondrial membrane fluidity, and (3) reduction in deleterious free radical activity.
Many nutrients play indispensable roles in mitochondrial energy production and provide vital antioxidant protection against the free radicals generated by oxidative phosphorylation. The substances in Table I have been shown to have generally positive effects on mitochondrial dysfunction-related conditions in a number of animal and human studies. These substances have alleviated a number of mitochondrial-induced diseases, including Parkinson’s disease, diabetes, and fatigue. A growing number of physicians and scientists believe that mitochondrial support nutrients may prove to be helpful for many of the symptoms associated with aging and age-related diseases.
References
1. Hutchin T and Cortopassi G. A mitochondrial DNA clone is associated with increased risk for Alzheimer’s disease. Proc Natl Acad Sci USA 92: 6892-95, July 1995.
2. Richter, C. Oxidative damage to mitochondrial DNA and its relationship to aging. Int J Biochem, Cell Biol, 1995,27:7, pp647-653.
3. Papa, S. Mitochondrial oxidative phosphorylation changes in the life span. Molecular aspects and physiopathological implications. Biochimica et Biophysica Acta, 1996),87- 105.
4. Cortopassi, G., and Wong, A. Mitochondria in organismal aging and degeneration, Biochimica et Biophysica Acta, 1999, 1410: 183-193.
5. Harman, Denham. The Biologic Clock: the Mitochondria? J Am Geriatr Soc, 1972,20: 145-147.
6. Miquel J, Economos AC, Fleming J and Johnson JE. Mitochondrial role in cell aging. Exp Gerontol 15: 575-91, 1980.
7. Miquel J. An integrated theory of aging as the result of mitochondrial DNA mutation in differentiated cells. Arch Gerontol Geriatr 12: 99-117, 1991.|
8. Miquel J. An update on the mitochondrial-DNA mutation hypothesis of cell aging. Mutation Research 275: 209-16, 1992.
9. Zs.-Nagy, Imre. A membrane hypothesis of aging. J Theor Biol, 1978,75: 189-195.
10. Zs.-Nagy, Imre. The role of membrane structure and function in cellular aging: a review. Mech Aging Dev, 1979,9: 237-246.
11. Zs.-Nagy, I., Ohta, M., and Kitani, K. Effect of centrophenoxine and BCE-001 treatment on the lateral diffusion and constant of proteins in the hepatocyte membrane as revealed by fluorescence recovery after photobleaching in rat liver smears. Exp Gerontol, 1989, 24: 317-330.
12. Zs.-Nagy, Imre. The horizons of an interdisciplinary synthesis in experimental gerontology. Arch Gerontol Geriatr, 1991, 12: 329-349.
13. Dean, W., Imre Nagy, Antiaging Pioneer, Vitamin Research News 2001: Vol. 15-2.
14. De Grey, A. The Mitochondrial Free Radical Theory of Aging, R.G. Landes, Austin, Texas, 1999.
15. De Grey, A. Reductive Hotspot Hypothesis of Aging, R.G. Landes, Austin, Texas, 2001.
16. Shigenaga MK, Hagen TM and Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 91: 10771- 78, Nov 1994.
17. Barrientos, A., Casademont, J ., Cardellach, F ., et al. Qualitative and Quantitative changes in skeletal muscle mtDNA and expression of mitochondrial-encoded genes in the human aging process, Biochemical and Molecular Medicine, 1997, 62: 165-171.



{kind=link}
[…] Though the mechanisms by which a high fat diet induces mitochondrial dysfunction is not clear, one can speculate that a diet high in fat is also low in critical nutrients that are required for proper mitochondrial function and cellular energy production. Deficits in nutrients such as thiamine (vitamin B1) can and do cause severe mitochondrial dysfunction and lead to an array of disease processes from nervous system destabilization to cardiac, GI and reproductive dysfunction. Other nutrients and cofactors are also critical for mitochondrial function. CoQ10, L-Carnatine for example, have been found helpful in mitochondrial induced dysautonomia, and the list goes on. […]
[…] there does seem to be some that may help with optimal mitochondrial function. Here are the main […]
[…] supplements that have been shown to increase mitochondrial number and function […]
[…] of energy required per cell. It is within this organelle that cellular respiration takes place to synthesize ATP (adenosine triphosphate), the energy molecule used in numerous body processes. The two main aerobic […]
[…] there does seem to be some that may help with optimal mitochondrial function. Here are the main […]
[…] English, James. Mitochondrial Dysfunction, Nutrition and Aging (September 21, 2013). Retrieved from https://nutritionreview.org/2013/09/mitochondrial-dysfunction/. […]
[…] The health of your mitochondria are of utmost importance in aging and age-related diseases. Tissues with the densest population of mitochondria are muscle and brain tissue, and this makes sense, considering our bodies’ energy expenditure is the highest in these locations. […]